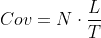I have performed RNA-seq analysis using HISAT2 & StringTie workflow suggested in: Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown.
Some of the transcripts/exons have decimal coverage values (eg., 1.1 or 2.59).
My question is: How can these tools report decimal coverage? If only half of the read overlaps exon will this exon have 0.5 coverage?