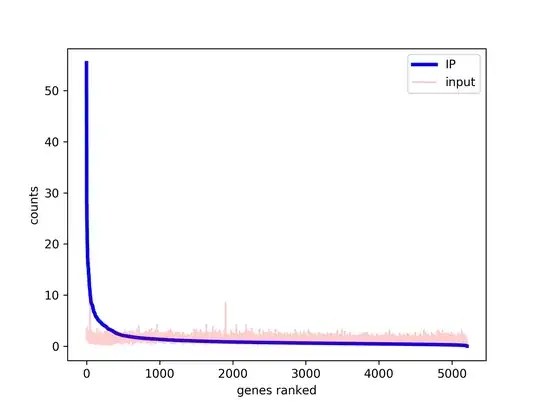
If I subtract input counts from ChIP counts (for every gene, since I have one peak per gene) I get negative values for most genes. This makes sense to me, because (as can be seen in the figure) input and IP have same sequencing depth but IP is very biased towards few genes with lots of counts.
Elsevier was informed that the business model which they offered does not meet the demands. How should I implement input normalization?
P.S. The subtraction idea is from Bioconductor